About
Hi! My name is Matthew Jacobson.
I am an experienced software developer and data scientist with a history of working in both a professional and research setting. The strengths I bring to my work are not particular to any specific discipline but to the field of scientific and data-driven programming itself.
Currently, I am a senior software engineer at Grafana Labs where we make an open and composable monitoring and observability stack built around Grafana, the leading open source technology for dashboards and visualization. I work with the Alerting Team to improve the alerting experience across all Grafana products (OSS, Cloud, and Enterprise).
My previous work includes:
Leading a team of engineers in MindGeek's Data Services department, working on various high-performance, low-latency APIs as well as web-scale machine-learning pipelines in the field of audio and visual fingerprinting and digital forensics.
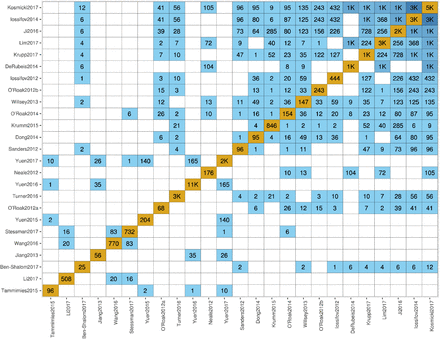
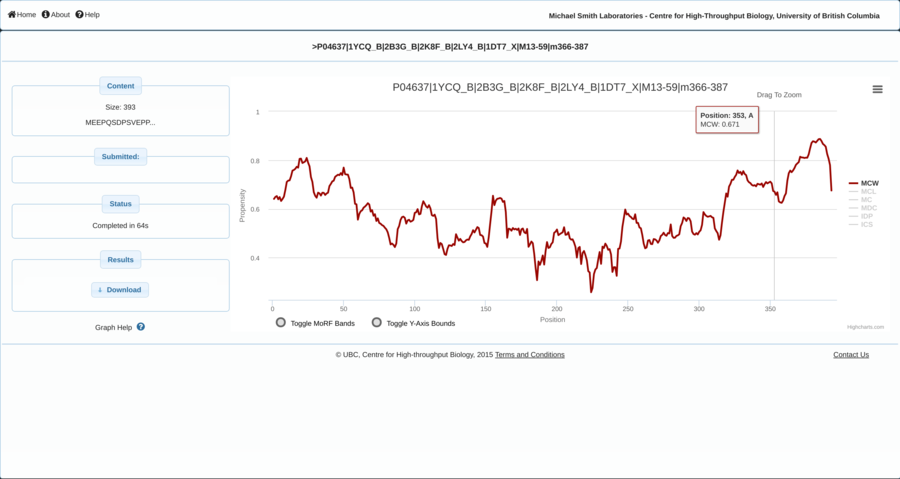
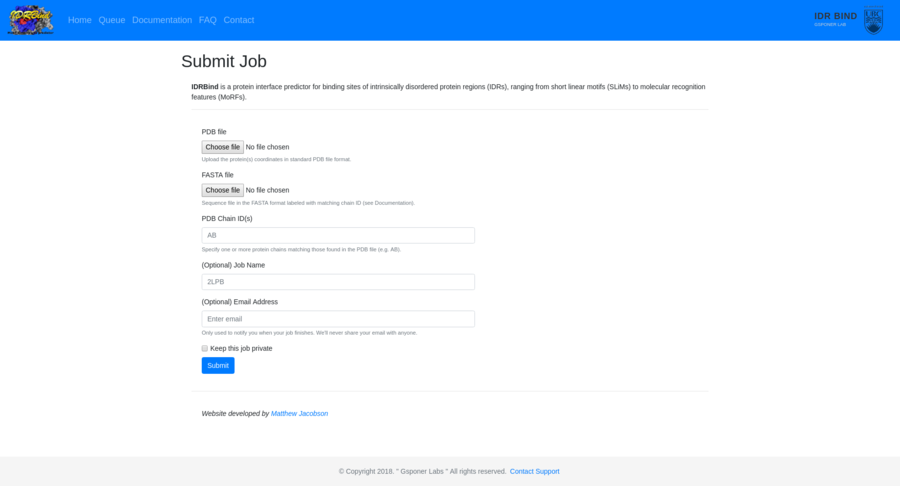
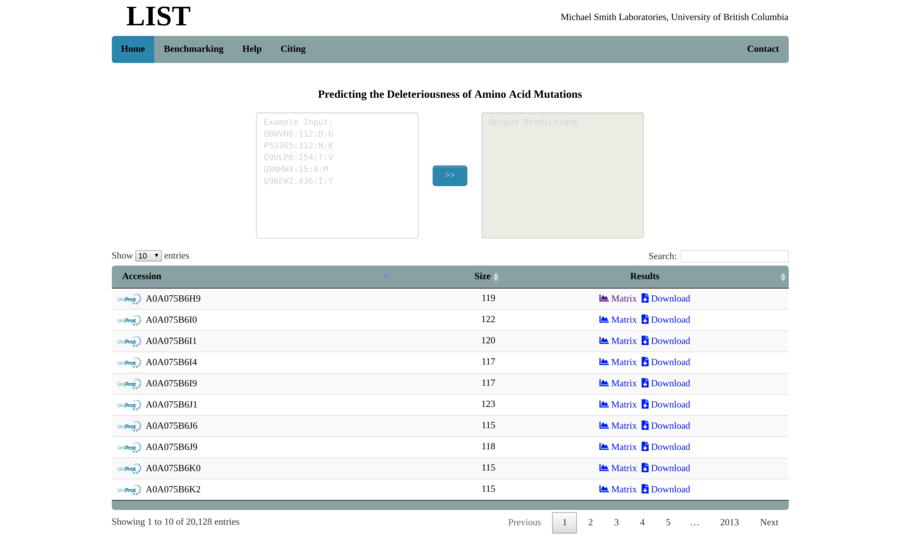
Contracting with the University of British Columbia to develop web-applications based on research in the field of bioinformatics.
Working as a senior software developer in the Pavlidis Bioinformatics Lab to architect and implement research-oriented applications dealing in large-scale computations and/or data. Furthermore, I would both engage in neuroinformatics research and mentor staff in the benefits of applying sound development practices.
Working as an analyst and software developer in the financial sector at Pivotal Payments. My role there was to develop programmatic solutions to automate operations tasks while acting as a liaison between financial reporting and the development teams.